The co-existence of autism spectrum disorder (ASD) and epilepsy is very much prevalent in certain neurodevelopment disorders and their association could be best understood by examining shared phenotypes, pathophysiological mechanisms and risk factors involved. Various genetic as well as environmental factors affecting the role of certain biological pathways in synapse formation and its maintenance along with cellular growth factors and regulatory proteins have been found to be responsible for the complexities involved in co- occurrence disorders. In this review, we have briefly described potential in vivo as well as in vitro models that have helped us in validating certain associated risk factors and decipher the bases of mechanism in developing both disorders, thus further helpful in finding useful target- specific therapy for patients.
Keywords:
Autism Spectrum Disorder; Co-Occurrence; Epilepsy; Genetic Models; Induced Models; Zebra Fish.
Abbreviation:
ASD: Autism spectrum disorder
TSC: Tuberous sclerosis complex
FXS: Fragile X syndrome
RTT: Rett syndrome
NRXN: Neurexins
NLG: Neuroligins
CAM: Cell adhesion molecules
CNTNAP: Contactin-associated proteins
nRT: Nucleus reticularis
mTOR: Mechanistic target of rapamycin
GABAA: Gamma-Amino Butyric Acid-A
VPA: Valproic acid
IL: Interleukin
EEG: Electroencephalogram
PMDS: Phelan-McDermid syndromes
ADDM: Autism and Developmental Disabilities Monitoring
MRI: Magnetic resonance imaging
FMRI: Functional magnetic resonance imaging
KO: Knockout
KI: Knockin
CDFE: Cortical dysplasia-focal epilepsy syndrome
PTZ: Pentylenetetrazole
iPSCs: Induced Pluripotent Stem Cells
ARX: Aristaless-related homeobox
CNS: Central nervous system
TRAPP: Transport protein particle
TALEN: Transcription Activator-Like Effector Nuclease
ZFN: Zinc Fingers Nuclease
Autism Spectrum Disorder and Epilepsy are two etiologically heterogeneous syndromes that are characterized by significant variability in clinical symptoms and often co-occur with other neurodevelopmental disorders such as developmental delay, intellectual disability, and behavioural deficits [1]. Epilepsy is a neurological condition that causes recurring seizures of various forms and up to 70% of epilepsies are estimated to have a strong hereditary basis due to genetic defects [2]. While, ASD is a broad category of neurodevelopmental disorders marked by difficulties with social interaction, nonverbal communication, and repetitive behaviour, all of which have various degrees of impairment [3]. Tuberous sclerosis complex (TSC), fragile X syndrome (FXS) and Rett syndrome (RTT) are some of syndromic types (of ASD) which present autism as one of the phenotypes and occurrence of epilepsy is nearly common in these disorders [4].
It has long been known that ASDs and various types of epilepsy (epilepsies) coexist and link between both disorders is very well-known, but elucidating the underlying mechanism is still a matter of concern. Because of the rising occurrence of ASD in epilepsy and epilepsy in ASD, each disorder is now regarded as a "comorbidity" of the other. Up to 30% of nonsyndromic autistic people experience seizures, and even more show epileptiform discharges on electroencephalogram (EEG) [5]. According to a systematic review by Lukmanji et al., the median overall period prevalence of epilepsy in autistic people was found to be 12% whereas, the prevalence of autism in epilepsy was 9%, which include all types of population [6]. Several studies have found that epilepsy can occur before or after the onset of ASD symptoms and ASD-epilepsy comorbidity is diagnosed usually in early childhood and adolescence, suggesting that both diseases share a neurobiological basis [7]. Genetic diseases include Phelan-McDermid syndromes (PMDS), FXS, TSC, RTT, and maternal duplications on chromosome 15q11.2-q13.1, which are characterised by both early-onset epilepsy and ASD symptomatology, provide a substantial evidence for this idea [8]. One of the hypotheses suggests that impairment of common neurodevelopmental processes highlighted by the relatively large number of genes associated with both disorders is the reason for the co-occurrence of epilepsy and autism [9]. The excitation/ inhibition balance theory proposes that neurodevelopmental abnormalities, particularly in the GABAergic and glutamatergic systems, cause an imbalance in excitatory and inhibitory brain circuits, resulting in the pathophysiology of both disorders [7].
Since exact mechanism involved in the pathogenesis of cooccurrence of both disorders is not clear and such condition has a significant impact on affected people, a better picture of the etiology of this common comorbidity is critical. In fact, the discovery of shared underlying mechanisms could lead to a new and improved treatment choices. In this review we have briefly described few experimental models explaining the possible mechanisms for co-occurrence that might highlight new targets for broader treatment purposes.
The prevalence of ASD in children with epilepsy or vice- versa has always been a controversial topic. As per Autism and Developmental Disabilities Monitoring (ADDM) network data (2002), about 16% co-occurrence of ASD- epilepsy have been reported [10], and according to other reports, nearly 20-25% children with ASD suffers from epilepsy [11]. According to a population-based study, 44 percent of children with ASD were later diagnosed with epilepsy, while 54 percent of children with epilepsy were later diagnosed with ASD [12]. Risk for epilepsy is higher in children with ASD and intellectual disability (21.5%) compared to those without comorbid intellectual disability (8%), [13] and these risks have increased in children with ASD and intellectual disability after the age of 12 years, [11] as confirmed by the meta-analyses.
The various symptoms are related to the individuals having epilepsy with ASD. Individuals who are diagnosed with epilepsy and ASD face complications in interacting with peers. Such individuals are showing greater unusual sensory interests as well as higher rates of repetitive object use [14]. Individuals displayed considerably more maladaptive behaviours associated to ASD who are diagnosed with both epilepsy and ASD [15] like decrease in communication, motivation and social cognition scores and also show increase levels of compulsive, self-injurious as well as sameness behaviours. Rates of impaired facial recognition and theory of mind were higher in patients with epilepsy and ASD compared to controls was concluded by a meta-analysis of social cognition in epilepsy and autism. Development of a child was normal until the onset of seizures at 21 months of age and by 24th month, the child met criteria for both ASD and ID, confirmed by one of the case studies of TSC [16]. Therefore, there exists a deep interaction between seizures, intellectual disability, ASD and the onset of seizures in developmental regression and ASD diagnostic symptoms. Children with epilepsy and ASD is showing lower score on social maturity scales, poor quality of life, and exhibit higher use of psychotropic medications [18,21], along with poorer social outcomes [20]. Studies have indicated that individual functioning is affected by the higher levels of cognitive impairments in adults followed from childhood, with seizure frequency [21]. Abnormalities in EEG and seizures are linked to the aberrant behaviour [22] in children with ASD but without diagnosed epilepsy.
ASD and epilepsy are largely preceded by structural abnormalities of the brain. Some evidence for a common pathophysiology for ASD and epilepsy came from brain imaging examinations. In both ASD and paediatric epilepsies, magnetic resonance imaging (MRI) investigations revealed altered grey and white matter volumes and atypical brain growth trajectories. ASD and epileptic disease expression have also been connected to anomalies of subcortical volumes, cortical thickness, surface area, and white matter microstructure. The brain regions including the frontal and temporal lobes tend to be the most impacted, but the parietal lobe is also frequently affected [23]. The malformations of the cortical development includes focal cortical dysplasia and heterotopias in both ASD and epileptic brain tissues [7]. A series of studies have revealed the possible interaction between preexisting focal cortical dysplasia and tissue damage in epileptic patients [25,28]. On the other hand patients with ASD are also more likely to have focal cortical dysplasia [29,34]. Thus the coexistence of epilepsy and ASD could be explained by an epileptic network in the brain of ASD patients that was activated by pre-existing focal cortical dysplasia. A recent retrospective case–control study by Fujimoto et al has shown that epileptic patients with focal cortical dysplasia were associated with higher ASD scores than healthy volunteers and the tendency for ASD was more in epileptic patients with type 1 focal cortical dysplasia than focal cortical dysplasia type 2 [34]. The histopathological examinations performed on postmortem epileptic and ASD brain tissue revealed focal cortical dysplasia characterized by the histological features including the radial dyslamination, minicolumn pathology, increased small diameter immature neurons, increased neurons in superficial white matter, indistinct gray-white matter boundary, ectopic neurons in the molecular layer and absence of balloon cells at various locations of brain including multifocal cerebral patches, frontal lobe, anterior cingulate, entorhinal cortex, cornu ammonis, dentate gyrus and cerebellur [36, 41]. Other histopathological findings common to ASD and epileptic brain includes heterotopia at subcortical, periventricular, hippocampus and cerebellum and increased dendritic spine density (pruning defect) at layer II of the frontal, temporal and parietal lobes and layer V of temporal lobe [42,43]. The discovery of structural indicators that occur before the onset of symptoms creates an ideal window for intervention. However, more studies are required to understand the particular properties of these biomarkers and their predictive usefulness.
The fact that ASD and/or epilepsy are etiologically heterogeneous disorders makes it difficult to properly anticipate diagnosis. Whole genome/exome sequencing has enhanced our understanding of the genetic roots of these disorders, and over half of all ASD and epilepsy cases are now known to have a genetic foundation [43]. Gene/environment interactions are recognised to have a role in the etiology of both disorders, and several environmentally induced models have been developed as well. However, no animal model can fully depict the involvement of either disorder; they can give an avenue for understanding the underlying cellular and molecular pathways.
Animal models have been extensively used to decipher the complex aetiology involved in neurological disorders as they are helpful in validating the role of various selected factors including genetics, environmental in a course of diseases. Moreover, animal models provide a valuable insight in examining the therapeutic efficacy of several new drug entities or repurposing the existing drugs against disease, as establishing a human neuronal cell cultures with its extensive proliferative capacity is a difficult task. In order to test a drug, a whole animal models recapitulating relevant human characteristics of autism and epilepsy both, which include pathology of disorders, construct validity in context to neurobiology, face validity, a significant analogies to patient’s phenotype and must respond to the treatment against the modelled disorder [45,46]. These models give us an indirect window into human pathophysiology, allowing us to discover new therapeutic targets and test the efficacy of treatments in preclinical studies. Despite inherent differences in human and mouse neurobiology at all levels of organisation, mice have been proved as an excellent models for human neurogenetic diseases. Over 99% of mouse genes have human homologues [46], and the spatial structure in the brain is similar and expression of genes is also conserved [47].
As of present information, both genetic and environmental risk factors are responsible for the co-occurrence of epilepsy and ASD. The evolution of genome editing system has helped in clarifying the role of genes and their relationship with phenotypes associated with a disease. CRISPR/Cas9, a programmable endonuclease-based genome editing system, has greatly simplified the establishment of clinically relevant genetic animal models [48]. Generating models of specific genetic modifications like complete knockouts (KO), knock-in (KI) and particular missense mutations using this technology has made it easy to understand an absolute outcome of a specific mutation. Following evidences based experimental rodent models will provide us an overview about the role of genetic mutation as a risk factor for development of ASD-epilepsy co-morbidity and are well summarized in Table 1.
Table 1: Genetic models of ASD-Epilepsy co-morbidity.
ASD-Epilepsy associated genes/model |
Name/role of gene product |
Gene modification technique |
Observed phenotype |
Reference |
CNTNAP2/Cntnap2 (Mice) |
Contactin associated protein-like 2; |
Targeted KO by gene replacement |
Abnormal EEG; Spontaneous seizures in >6 month old animal; |
[22] |
NLGN2/Nlgn2 (Mice) |
Neuroligin 2; |
- |
Abnormal EEG; |
[19] |
SCN1A/Scn1a (Mice) |
Sodium voltage-gated channel,alpha subunit 1; |
Target deletion of exon |
Hyperactivity; |
[9,71] |
SCN2A/Scn2a (Mice) |
Sodium voltage-gated channel, alpha subunit 2; |
- |
Abnormal EEG; Stereotyped repetitive behavior; Behavioral arrest |
[10] |
SCN8A/Scn8a (Mice) |
Sodium voltage-gated channel, alpha subunit 8; ion channel |
R1620L mutation by CRISPR/Cas9 technology |
Hyperactivity; |
[11] |
KCNQ2/kcnq2 (Mice) |
Potassium voltage-gated channel subfamily Q member 2; ion channel. |
KO by deletion of gene |
Repetitive behavior; |
[12] |
GABRB3/gabrb3 (Mice) |
GABAA receptor alpha 3 subunit; neurotransmitter receptor |
Targeted KO (Embryonic stem cells) |
Abnormal EEG; Seizures; Hyperactivity; Learning and memory deficits |
[13] |
GABRA4/gabra4 (Mice) |
GABAA receptor alpha 4 subunit; |
KO by transcription activator-like effector nucleases (TALEN) technology |
Autistic-like behavior; |
[14] |
SYN/SynI,II (Mice) |
Synapsins; regulate neurotransmitter release at synapses |
KO by Homologous recombination |
Impaired social interaction, Novelty and recognition; Epilepsy |
[25] |
EN2/En2 (Mice) |
Engrailed2; transcriptional regulator |
KO by targeted deletion |
Deficits in social behavior; |
[72,28] |
IQSEC2/Iqsec2 (Mice) |
IQ motif and Sec7 domain 2; |
KO using CRISPR/Cas9 technology |
Hyperambulation; Hyperanxiety; |
[29] |
FMR1/Fmr1 (Mice) |
Fragile X mental retardation 1; |
- |
Repetitive behavior; |
[90] |
MECP2/Mecp2 (Mice) |
Methyl CpG binding protein 2; |
KO by Zinc fingers nucleases (ZFNs) |
Aggression; |
[30] |
PTEN/Pten (Mice) |
Phosphatase and tensin homolog ; |
KO by Cre technology |
Hyperactive; |
[62] |
TSC1/Tsc1 (Mice) |
Tuberous sclerosis complex 1; |
Homologous recombination |
Autism-like behavior; |
[58] |
TRAPPC6B/trappc6b (Zebrafish) |
Transport protein particle; regulates membrane trafficking through the Golgi apparatus |
Knockdown by morpholinos (MOs) |
Decreased head size; |
[66] |
KCNJ10/kcnj10a (Zebrafish) |
Inwardly-rectifying potassium channels Kir4.1; |
Knockdown by morpholinos (MOs) |
Altered development |
[67] |
A highly organised protein complex consisting receptors, signalling molecules, and scaffolding proteins is essential for synapse formation. Cell adhesion molecules (CAMs) that are synaptically localised not only mediate a physical connection between cells, but they are also dynamic regulators of synapse function [49]. The interaction between presynaptic neurexins (NRXN) and their postsynaptic partner neuroligins (NLG), which act as calcium-dependent cell adhesion molecules, is well-studied trans-synaptic signal involved in synaptogenesis [50]. Studies have reported that individuals with genetic mutations in NRXN1 (deletions) [51] and NLG [52] are found to be affected with ASD, schizophrenia, intellectual disability. Mutation in contactinassociated proteins (CNTNAP) has been found to be associated with ASD and cortical dysplasia-focal epilepsy syndrome (CDFE) and it has a role in axonal and dendritic molecular organizations. Results obtained from the study of Cntnap2 KO mice have also supported these evidences by presenting phenotypes of both disorders [53].
Nlg2 KO mice had reported an aberrant EEG activity and behavioural arrests that are similar to absence seizures due to the impaired GABAergic transmission in nucleus reticularis thalamic (nRT-thalamic) circuit which represent a similar mechanism underlying both epileptic seizures and ASD [54]. A study has reported that a point mutation (R451C) has resulted in ASD in humans, which is further confirmed by Nlgn3 KI mice model by showing abnormalities in social interaction, which is implying a gain-of-function effect for this mutation. The fact that this model was resistant to seizures caused by pentylenetetrazole (PTZ) suggested a change in GABAergic inhibitory neurotransmission in the NL3R451C (Neuroligin3- R451C) mices’ cerebral cortex [55]. A high majority of occurrences of ASD are likely to be caused by genetic mutation of the SHANK3 gene, which is a scaffolding protein essential in the maintenance of the postsynaptic density in excitatory synapses. Shank3b KO mice showed a significant resistance to PTZ seizure induction as well as an increase in gamma band oscillatory EEG activity, which indicates increased inhibitory tone. Shank3b KO mice have also shown repetitive grooming, social interaction deficits along with decreased open field activity, as well as low in sensory, learning, memory and anxiety-related behaviours [56]. Although these above-described models give significant construct validity but behaviour, representing both phenotypes in single model has not been reported by some studies. Thus, further characterization of these models is required to validate them.
The fact that ASD and epilepsy are co-occurring frequently suggests that they are the outcome of a shared underlying neurological mechanism. The abundance of rare genetic variants reported in autism and epilepsy that indicate the presence of impairments in both synapse formation and function, generating a disrupted balance between neuronal excitation and inhibition, supports this perspective [4]. Ion channels, transmembrane protein complexes that undergo structural changes to allow the passage of ions across the neuronal membrane, are responsible for determining neuronal excitability. Seizures and epilepsy are triggered by a disruption in the balance between neuronal excitation and inhibition (E/I balance), where dysfunction in sodium or calcium channels triggering the depolarisation of membrane has been linked to enhanced excitation while the mutations in potassium channels is related to the inhibitory functions. Comorbid ASD and epilepsy have been linked to genetic abnormalities in genes that encode both voltage and ligand-gated ion channels [57].
Experimental models expressing the mutations in voltage gated sodium channels, NaV1.1 [58], Nav1.2 [59] and Nav1.6 [60] encoded by Scn1a, Scn2a and Scn8a respectively reportedly mimicked the phenotypic profile of autism and epilepsy such as hyperactivity, spontaneous seizures, stereotyped repetitive behaviour, impaired spatial memory, learning and social deficits etc. Similarly, mice showing loss of function mutation of voltagegated potassium channel subunit Kv7.2 encoded by Kcnq2 also displays ASD symptoms and associated epileptic behaviour [61]. Models displaying mutations in ligand-gated channels such as mice model of Gabrb3, which encodes for the β3 subunit of the Gamma-Amino Butyric Acid-A (GABAA) receptor [62] and Gabra4 KO C57BL/6 mice, have also presented a phenotype of both epilepsy and autism [63].
Synapsins (Syns) are a family of synaptic vesicle (SV) phosphoproteins, which are encoded by the SYNI, SYNII and SYNIII genes [64]. The role of SYN1 gene (on X chromosomes), associated with cytoplasmic surface of synaptic vesicles has been found to play a critical role in synaptogenesis and regulation of neurotransmitter release. Recent findings have suggested that individuals with nonsense mutations in SYN1 are more susceptible to ASD and epilepsy [65]. Mice model with mutation in Synapsin (SynI−/−, SynII−/−, SynIII−/−) has presented ASD- epilepsy related phenotypes implying that dysregulation in synaptic homeostasis might be one of the majors factors responsible for the development of co-occurrence of diseases [66].
For appropriate brain development, control of protein synthesis at various developmental stages is essential and co-occurrence of ASD and epilepsy are outcomes of the mutations in genes that encode regulatory proteins involved in mRNA translation, DNA transcription, and activation of protein. Transcriptomic profiles have revealed that mutations in various genes involved in the regulation process in ASD-epilepsy comorbidity after analyzing the mRNA expression of hippocampus, cerebral cortex and cerebellum at several embryonic and postnatal stages [7]. Dysregulation in some of the transcription factors like Aristalessrelated homeobox (ARX), ENGRAILED 2 (En2) and IQ motif and Sec7 domain (IQSEQ2) have been reported to be involved in epilepsy-ASD comorbidity. The ARX gene encodes for a pairedtype homeodomain transcription factor that plays an important function throughout embryonic development. Transcriptome study of embryonic (E12.5) telencephalon from ARX mutant mice has revealed many dysregulated genes associated with ASD and epilepsy [67]. Latest human genetic studies have suggested that the EN2, a homeobox transcription factor involved in pattern formation in developing CNS, have been implicated in cooccurrence of ASD and epilepsy. This is further supported by the animal study in which En2 KO mice have reported phenotypes like neurobehavioural abnormalities in social, learning, and memory activities [68]. Also, transcriptomic data of adult hippocampus of En2 KO (En2 -/-) mice have illustrated a significant enrichment of differentially expressed genes including ASD and epilepsy genes [7]. In addition to these, the role of X-linked gene, IQSEQ2 on Chromosome Xp11.22 has been found to be related with multiple cases of ID, ASD, epilepsy and epileptic encephalopathy. Iqsec2 deficient mice have presented a phenotypic profile of both disorders like hyperambulation, hyperactivity and deficits in growth [69].
Role of fragile X mental retardation 1 (FMR1) gene, located at Xq27.3, has always been critical in regulating the normal cognitive development and several other important processes. Mutations in FMR1 have been identified as a largest contributor to ASD [70] and associated increased susceptibility to other medical conditions like seizures, recurrent otitis media etc. [71]. In fact, Fmr1 KO mice have presented symptoms of ASD like repetitive behavior and hyperactivity and were found to be susceptible to seizures [72]. The methyl CpG binding protein 2 (MECP2) gene, located on Xq28, is well known for its role in regulating human and mouse brain morphology, which acts as a transcriptional repressor that binds to the methylated CpG dinucleotides. Tremors, anxiety-related behaviours, and aberrant seizure-like brain activity were observed in mice lacking Mecp2 (KO) in their glutamatergic neurons [73]. Dysregulation of RBFOX, a neuronal splicing regulator, has been found to be implicated in various neurodevelopmental disorders such as ASD, epilepsy, cognitive impairments etc. RBFOX3/NeuN is a member of the RNA-binding Fox (Rbfox) family of proteins, which regulates alternative RNA splicing and is particularly expressed in neurons. Rbfox3 homozygous KO mice exhibited impaired cognitive abilities (learning deficits) due to deficits in neurogenesis and also displayed cold hyperalgesia justifying the phenotypic profiles of both disorders [74]. However, this model explains more about mechanistic role leading to these phenotypes and fails in construct validity.
Several other genetic mutations have affected various aspects of neuronal functions which are discovered to be associated with development of ASD and epilepsy. Indeed, such mutations affect proteins involved in all aspects of neuronal excitability, which involve anchorage of synaptic circuitry, management of synaptic vesicle release, controlling subcellular signaling pathways, regulating neuronal migration, and organising network connections [4]. Dysregulation in these cellular factors can bring alteration in formation of circuit and their transmission. Mechanistic target of rapamycin (mTOR) pathway plays a critical role in synthesis of synaptic proteins and integration of inputs from receptors like N-methyl D-aspartate (NMDA) and metabotropic glutamate [57]. mTOR is a serine/threonine protein kinase that is involved in two distinct protein complexes, mTORC1 and mTORC2, which promote cellular growth and survival, respectively [75]. Alteration in the activation of mTOR pathway is usually found in syndromes like tuberous sclerosis and Cowden syndrome due to mutations in TSC1, TSC2 [76] and Phosphatase and tensin homologue on chromosome 10 (PTEN) [77]. Surprisingly, epilepsy is the most frequent neurological condition associated with tuberous sclerosis, accounting for 80–90% of cases, and around 50% of tuberous sclerosis patients also have ASD [76]. TSC-mTOR signaling regulates protein synthesis, which is involved in learning-related synaptic alterations. Apparently, one of the studies have demonstrated that model of TSC resulted in epilepsy-induced mTOR hyperactivation in raphe nuclei promotes autism like behaviour [78,79] Pten deficient mice have been found to be deficits in social interaction and marble burying [79] and also ASD and epilepsy-related symptoms have been reported in conditional deletion of Pten in pyramidal neurons by using Cre technology [80].
When healthy animals are made to develop a desired disease model either by exposing them to a chemical compounds at postnatal stages or during prenatal stage, are called induced models and they have been used for over 75 years. They have been proved very helpful in identifying a particular disorder and developing a treatment for the same. In order to explain the mechanism responsible for the co-occurrence of ASD and epilepsy, here are explained few animal models showing the phenotypes of both disorders and briefly summarized in Table 2 as well.
Animal models of seizures induced by chemicals are commonly used to investigate the pathology involved in epilepsy. Moreover, use of chemicals for induction has also proven helpful as timing and number of seizures could be easily controlled. Of them, pentylenetetrazole (PTZ), a GABAA receptor antagonist, is being used to generate acute seizures to severe tonic-clonic seizure in animals, depending on the dosage [81]. PTZ-kindled mice model of epilepsy has reported to display a phenotype profile of both disorders like impaired motor coordination, anxiety, and social approach impairment. This study suggested the fact that α4 nicotinic receptor down regulation and NLG3 up regulation in the piriform cortex are closely related to the ASD- epilepsy comorbidity [82]. Valproic acid (VPA), an epileptic drug, is reported as a well-known environmental risk factor responsible for ASD in humans and animal model of VPA has also exhibited autistic like features along with the increased susceptibility to seizures and hyperactivity [83].
Table 2: Induced model of ASD- Epilepsy co-morbidity.
Induced chemicals/ Environmental risk factors |
Observed phenotype |
Reference |
PTZ (Mice) |
Impaired motor coordination; |
[32] |
VPA (Mice) |
Increased seizure susceptibility, Hyperactivity; |
[73] |
PIC (Mice) |
Impaired social interaction; |
[34] |
Maternal stress and terbutaline (ST model) (Rat) |
Autistic-like behaviour ; |
[35] |
Abbreviations: PTZ: Pentylenetetrazol; VPA: Valproic acid; PIC: Polyinosinic–polycytidylic acid. |
||
One of the major environmental risk factors responsible for ASD is immune activation caused by maternal infection during pregnancy, where immunogens like polycytidylic acid (poly (I:C) and lipopolysaccharide (LPS) are being given to mimic the viral and bacterial infections, respectively [84]. Of them, mice administered with PIC during pregnancy displayed impaired social interaction along with faster progression of seizures and increased hippocampal excitability. The study also suggested the major role of IL-6 and IL-1β in the mechanism underlying the co-occurrence of autism and epilepsy [85]. The combination of maternal stress and terbutaline (tocolytic β2 adrenergic receptor agonist) has been found to cause autistic-like behaviour along with spontaneous recurring convulsive seizures in 45% and epileptiform spikes in 100% offspring of rats due to hippocampal gliosis [86].
Using cellular models for understanding a disease has always been a great help in modeling disorders with complex mechanisms involved. By expressing four genes known as the Yamanaka factors (Oct3/4, Sox2, Klf4, and c-Myc), these approaches can turn differentiated cell lines into induced pluripotent stem cells (hiPSCs), which was first generated in 2007. Human induced pluripotent stem cells (iPSCs) can now be produced efficiently from both neurological patients as well as healthy controls [87] and mutations present in single gene disorders can be recapitulated by editing control iPSC lines using CRISPR/Cas9 tools [88]. These models are useful not only for establishing a genotype-phenotype association, but also for establishing novel therapeutic techniques, such as cell therapy and pharmacological treatments. Generally 2D monolayer culture models are being used to generate disease-relevant cell types in iPSC based modeling research studies. In several neurological disorders showing co-occurrence of ASD and epilepsy such as Tomothy syndrome (TS) [89], TSC [90], FXS [91], PMDS [92] and RTT [93], different iPSC-derived phenotypes in neural progenitor cells (NPCs), neurons, and glial cells (mostly astrocytes and oligodendrocytes) have already been documented.
The role of dysregulation of various important signaling pathways suggests to contribute in the pathogenesis of co- morbidities of diseases has always been the matter of concern. Similarly, impairment in some of the pathways involved in protein translation regulation, such as mTORC1 [94], MEK-ERK, and MNK-elF4E [95], are linked to the pathogenesis of ASD and epilepsy comorbidity, and this has also been confirmed using iPSC-derived NPCs from patients with specific mutations and phenotypic profiles of both disorders. Vast number of research in this field is being conducted worldwide to explore and elucidate the genes associated in ASD and epilepsy comorbidity using this approach.
Furthermore, with recent advancements, it is now possible to differentiate single iPSC cell lines and arranged them into threedimensional (3D) organoids, offering a model that more closely reflects tissue and organ-level disease pathophysiology and mimics the intricacy of the brain's architecture. These organoids can summarise sequential neurogenesis, migration, gliogenesis, and synaptogenesis of cortical development, which cannot be investigated in 2D cell cultures [96]. Also, organoids are made up of a variety of neuronal and glial cell types that have limited self-organisation and spontaneous, synchronised neural activity, implying that they could be used to model both network-level and cell-autonomous defects involved in both disorders [97]. Similar studies have been performed by Blair et al., using organoid reflecting TSC phenotype made up by combining CRISPR/Cas9 genome editing and brain organoids, suggesting role of impaired mTORC1singalling pathway in pathophysiology of ASD-epilepsy comorbidity [98]. In vitro models associated with ASD-epilepsy co-morbidity are summarized in Table 3.
Table 3: In vitro models explaining the co-morbidity of ASDEpilepsy.
hiPSCs- based models |
ASD-Epilepsy associated mutated gene |
Key findings due to altered gene expression |
Reference |
Neural progenitor cells |
ARHGEF9 |
Altered mTORC1 signalling pathway |
[43] |
TSC2 |
mTORC1 activation; |
[44] |
|
Cortical neurons |
MECP2 |
Alteration in neural network synchronization; |
[74] |
NRXN1 |
Altered calcium dynamics; |
[75] |
|
TSC2 |
Hyperactivation of mTORC1; |
[76] |
|
Brain organoid |
TSC1 |
Impaired developmental suppression of mTORC1 signaling by loss of either TSC1 or TSC2 |
[47] |
Abbreviations: mTOR: The mechanistic target of rapamycin |
|||
Considering all the results, it is clear that despite the benefits of in vitro models, cell culture cannot properly reflect all of the complexities involved in the development of ASD and epilepsy; as a result, animal models remain a critical tool for fully comprehending them.
The zebra fish (Danio rerio) ) are freshwater teleost fish, has been proposed as an effective animal model for studying the genetic background of various human neurological disorders such as intellectual disability, epilepsy [99] and ASD [100] in recent years. The zebrafish has also been represented as a robust model for studying several diseases and shows significant genetic homology (approximately 70– 80% of orthologous genes) with humans [101]. It is easy to generate mutations in their genomes by using CRISPR methods impacting any gene of interest (GOI). However, it is still difficult to a mimic a phenotype of a particular disorder in mutant larvae. Role of several mutations generating ASD and epilepsy in patients have been confirmed using zebrafish models.
Transport protein particle (TRAPP) is a multisubunit complex that regulates membrane trafficking through the Golgi apparatus. Patients with homozygous splice mutation in TRAPPC6B presented autistic and epileptic feature and zebrafish trappc6b morphants generated by using morpholinos (a translation blocking and splice blocking) displayed a decreased head size, neuronal hyperexcitabililty with lower threshold to seizure when treated with PTZ [102]. As already mentioned earlier, dysfunctioning of the Kir4.1 (encoded by gene KCNJ10) represents a pathogenic mechanism contributing to ASD- epilepsy comorbidity, which has also been explored using kcnj10a morphant overexpressing the mutated human KCNJ10. The abnormalities in organ development and neuronal hyperexcitability underlying epilepsy were observed in morphants, implying the role of gain of function defects of astrocytic Kir4.1 channels in the pathophysiology of ASD- epilepsy comorbidity [103]. Since this model is easily available for screening different types of drugs as a treatment against various disorders, further investigation is necessary in this regard.
The focus of present review is to improve our knowledge and understandings of complex genetics involved in the pathogenesis in ASD-epilepsy comorbidity. Considering all the evidence, it is required to develop a reliable model that could be used for the discovery of potential therapies. Animal models enable us to explore and understand the pathophysiological mechanisms and can be manipulated genetically for evaluating the impact of proposed treatments, despite the fact that face and construct validity are inherently imperfect. By using CRISPR/Cas9 genomic editing tool, it is now possible to reconstruct a particular mutation in models that are found in patients. In spite of the potential available models for neurodevelopmental disorders, there are still some lacunae that are needed to be resolved as currently no model is presented as ‘bestfit’ that could explain the cooccurrence of ASD-epilepsy.
Developing an animal model of any disease, especially those involved with complex etiologies is challenging. However, autistic-like phenotypes could be predicted by quantifying behavioural parameters, it is still difficult to ascribe “autism” to animals. Therefore, only human could be the best model for autism, as modeling autism in animals could only meet face and construct validity that will advance translational research. Likewise, co-occurrence of ASD and epilepsy cannot be explained purely on the basis of epileptic phenotype shown by above mentioned animal models and alternative models of epilepsy (status epilepticus, temporal lobe epilepsy model, etc.) could be considered and extensively studied to get an idea of epileptic synaptic phenotype.
Considering in vitro studies, where iPSCs as a tool has somewhat uncovered various mechanisms at cellular and molecular level underlying ASD-epilepsy co-morbidity and enabled the progress in therapies. Reported studies have provided us with the evidences of shared genetic vulnerability in the ASD–epilepsy connection (Figure 1). Mutations in several genes involved in the neuronal migration, synaptic formation and transmission etc., have found to be associated with development of ASD and consequently epilepsy or vice-versa. Moreover, E/I imbalance resulting in hyperexcitability has also suggested to contribute to ASD-epilepsy comorbidity. However, these ideas need a wide exploration for reaching out to new targets as therapeutic approach against such condition.
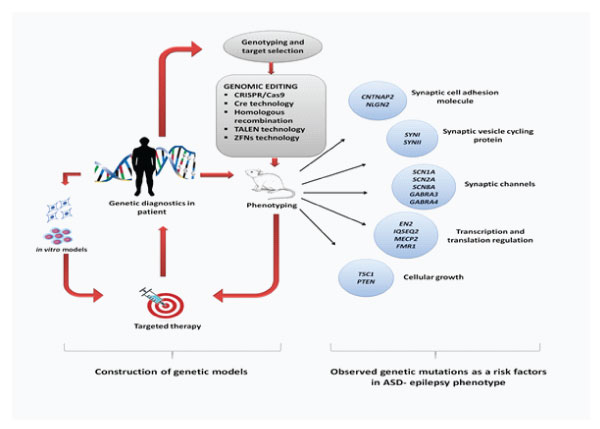
Figure 1: Schematic representation of experimental models (in vitro and in vivo) used to understand the mechanism behind the co-occurrence of ASD and epilepsy along with the genetic mutations observed in co-morbidity. Genetic variants diagnosed in patients can be well studied by using iPSC-derived cells from those suffering with specific mutations and showing phenotypic profiles of both disorders. Similarly, certain mutations presenting as a risk factors in ASD-epilepsy comorbidity could be well understood by using genetically manipulated animal models reflected through their phenotypes, which further provide a platform for mechanistic understanding and development of new target specific therapies in patients.
All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
None declared.
The authors declare no conflicts of interest regarding this article.