We wish to present a very unusual case of an inheritance of a very rare disease, familial complete androgen insensitivity syndrome, in a baby girl with three mothers and the mother being a known case of complete androgen insensitive syndrome. A 6-week-old term apparently female baby girl was born by normal spontaneous vaginal delivery with a birth weight of 3200 gm. Physical examination showed apparently female phenotypically. Abdominal examination confirmed right inguinal hernia with palpable gonad at internal inguinal ring. Trans-abdominal ultrasound confirmed absence of any female pelvic organs and karyotype of the baby was 46 XY. The baby underwent examination under anesthesia, vaginoscopy, laparoscopic bilateral gonadal biopsies and bilateral inguinal hernia repair uneventfully and discharged home same day evening. Both gonadal biopsies confirmed them to be testes. The parents were very happy and it was nothing new for them, as they now know how to resolve the challenges and threats associated with this rare disorder of the sexual development.
Keywords: Complete Androgen Insensitivity Syndrome, Female Bilateral Inguinal Hernia, Gonadal Biopsy, Laparoscopy, Vaginoscopy, X-Linked Recessive Inheritance
Complete androgen insensitivity syndrome (CAIS) occurs when the body cannot use androgens at all. Patients with the CAIS have the external sex characteristics of females, but do not female pelvic organs like a uterus or ovaries and therefore do not ovulate, menstruate and are infertile . Recent advances in assisted reproductive technology (ART), coupled with the emerging understanding in molecular mechanisms of disorders of sex development (DSD) and that of associated genetic infertility, have given hopes for fertility in groups of patients who till recently were denied biological parenthood [1]. We wish to report an unusual mode and model of inheritance of CAIS from a mother to a baby who had three mothers as the original mother had CAIS so the oocyte was donated by her sister and the surrogacy was provided by her sister-in-law and the baby developed right inguinal hernia and detailed investigations confirmed her to be a case of CAIS. The aim is to report complication of our own managed case and the objective is how to avoid such problems in the future.
An apparently healthy 3200 grams weight full term baby girl was delivered via spontaneous vaginal delivery. Everything seemed normal till her first month when mum and dad came in to the general practitioner saying that they noticed a lump in her right groin when she was crying. There is no lump when she is sleeping or comfortable and noticed it on straining or crying.
This baby girl had three mothers- mother had hernia as a child and was operated as a baby and was told that she has CAIS and had no female pelvic organs so can’t ovulate, menstruate and will be infertile. Her junior sister donated egg and baby was carried by her husband’s senior sister as the surrogate. Parent's dream has now come true and they are mother and father to baby. In essence it was the demonstration of girl powers three as mothers and one as the baby and concerns were the daughter shows mother!
Parents were now concerned if their baby would be having the same syndrome. Although the egg was donated by her sister, she said that her sister was a known carrier and no tests were done on father and the baby as her sister-in-law who carried the baby was fine and can’t take part in the fertilisation of sperm and egg as a surrogate mother. They approached their general practitioner for concerns about the right inguinal hernia and for very rare possibility of recurrence of the same syndrome.
Referring general practitioner cannot see any hernia but the symptoms, storey and the lump position described by the parents suggest a hernia without any signs of strangulation or infection and wanted us to review the baby in view of the hernia and the possibility of testing for possible CAIS.
On physical examination, the baby appeared apparently female, active, pink and beautiful. Patient’s vital signs were within normal limits. Abdominal examination showed a lump coming up on crying in the right inguinal deep internal ring area and travelling down to groin till pubic tubercle. On palpation a firm mobile lump could be felt at the deep ring area which was disappearing deep into the abdomen on the soap test and appeared as a gonad. Her perineal examination revealed three openings-urethral, vaginal and anus in normal anatomical position, size and shape.
In view of the family history of CAIS, known mutation, inguinal hernia with a palpable gonad and parental and general practitioner concerns of very rare but realistic possibility of the recurrence of the same CAIS, the patient had an abdominal ultrasound and blood chromosome karyotype
Abdominal ultrasound demonstrated absent uterus and two gonads were seen close to the internal inguinal ring opening and no cervix could be identified but blind ending vagina could be seen. Genetic blood chromosome studies indicated karyotype 46XY. An apparently normal XY chromosome complement and banding pattern seen in peripheral blood cultures from this phenotypically female patient. In view of the reported family history, this finding was likely to be the result of CAIS. Molecular studies to confirm the presence of known familial mutation was recommended but refused by the parents in view of their short budget. It was suggested to get genetic counselling at an appropriate age as the development of germ cell cancer called gonadoblastoma may be a complication later in the life in CAIS.
A laboratory study of any genetic mutation of AR gene was carried out. Patient underwent examination under anesthesia and vaginoscopy which confirmed blind ending vagina and normal looking external female genitalia. Laparoscopy confirmed bilateral inguinal hernias and both gonads appeared like testes. Biopsies from both gonads were taken and bilateral laparoscopic repair of the inguinal hernias carried out.
The post-operative period was uneventful and the patient was discharged home the same day in the evening. Both gonadal biopsies confirmed them to be testes. At follow up, the results and implications were discussed with the parents. Both parents accepted this situation quite well and in fact they told us that it is nothing new as before the fertility advanced option full karyotype of the husband was suggested but it was not going to make any change in their situation and hence it was not done and her sister already was a known carrier but now they are experienced patients and parents and for them it was nothing knew as they who know how to deal with the problems of fertility in CAIS and accepted the situation as it was with the smile on their faces.
CAIS is inherited in an X-linked recessive pattern in which the mutated gene is located on the X chromosome. In genetic males with only one X chromosome, one altered copy of the gene in each cell is sufficient to cause the condition. In genetic females with two X chromosomes, a mutation must be present in both copies of the gene to cause the CAIS. Males are affected by X-linked recessive disorders much more frequently than females (Fig 1A). About 66% of all cases of CAIS are inherited from mothers who carry an altered copy of the androgen receptor (AR) gene on one of their two X chromosomes. The remaining cases result from a new mutation that can occur in the mother's egg cell before the child is conceived or during early fetal development [2].
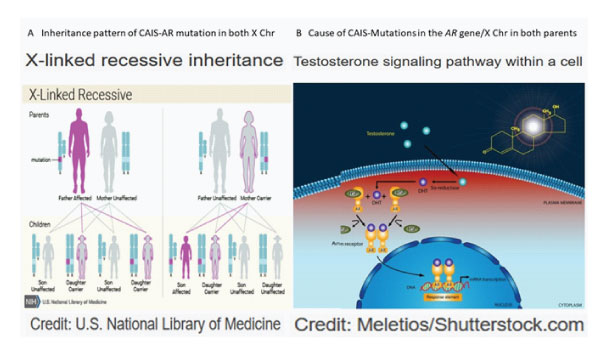
Figure 1: A: Inheritance pattern of CAIS and AR mutation in both X chromosomes. B: Cause of CAIS-mutation of AR gene on both x chromosomes and intracellular pathway.
CAIS is caused by mutations in the AR gene and is inherited in an X-linked manner The mutations in the AR gene, which provides instruction for making a protein called AR. The receptors are present in many of the body's tissues, where they bind to androgens. The resulting androgen-receptor complex then binds to DNA and regulates the activity of androgen-responsive genes. By turning the genes on or off as necessary, the androgen receptor helps direct the development of male sexual characteristics before birth and during puberty and also have other important functions in both males and females, such as regulating hair growth and sex drive. In one region of the AR gene, a DNA segment known as CAG is repeated multiple times. This CAG segment is called a triplet or trinucleotide repeat (Fig 1B).
Recent study showed that a novel mutation of the AR gene can cause CAIS; the study also broadened the AR mutation spectrum and indicated that targeted exome sequencing could help facilitate the diagnosis of complicated DSDs [3]. At the molecular level, this hormone resistance is caused by hemizygous loss of function mutations in the X-chromosomal AR gene [4]. More than 600 different mutations in the AR gene have been identified in CAIS. Most of these mutations are changes in single DNA building blocks called base pairs. Other mutations insert or delete multiple base pairs in the gene or affect how the gene is processed into a protein.in CAIS.
In CAIS, the penis and other male body parts fail to develop despite being 46 XY genetic sex. At birth, the child phenotypically looks like a girl with blind ending vagina and absent internal female genital organs. No precise figures are available for the prevalence of CAIS, but estimates range from one in 20 400 to one in 99 100 genetic males on the basis of a proven molecular diagnosis [1]. They are typically raised as females and have a female gender identity. They have undescended testes that are abnormally located in the abdomen/ pelvis as male internal sex organs with or without an associated inguinal hernia. People with CAIS also have sparse or absent hair in the pubic area and under the arms.
A female adolescent with the CAIS has breast development and a pubertal growth spurt at the appropriate age, but no menses. Development of oestrogen-dependent secondary sexual characteristics occurs as the result of excess aromatisation of androgens. Pubic and axillary hair is usually absent or can be present in sparse amounts Some CAIS patients, therefore, do NOT need the estrogen substitutive therapy since the androgen can transform into estrogen and maintain their secondary sexual characteristics, so they refuse to undergo gonadectomy. Our patient’s mother herself was a living example that she did not require estrogen substitution therapy despite removing both of her gonads early in childhood and still was able to maintain her secondary sexual characteristics. We, therefore, endorse this choice fully.
CAIS mainly presents as primary amenorrhea in an adolescent female or as a bilateral inguinal/labial hernia containing testes in prepubertal children. Some issues in CAIS such as the follow-up of intact testes, the timing of gonadal removal and optimal hormone replacement therapy remain poorly standardised. Basic research led to the new issues to improve long-term well-being such as bone health, immune and metabolic aspects and cardiovascular risk. An expert multidisciplinary approach is mandatory to increase the long-term quality of life of women with CAIS [5].
With regards to the development and the potential surgery or other treatment of vagina in CAIS patients, treatment and gender assignment can be a very complex issue, and must be individualized with each affected person. Most cases of CAIS will have adequate size of the blind ending vagina at birth and may grow as they approach puberty as has happened in our index case but some of them may not have adequately sized blind ending vagina. Vaginal length can be measured in prepubertal girls undergoing inguinal hernia repair to screen for CAIS and a shortened vagina and the absence of ovaries or fallopian tubes suggests the need for karyotyping. The uterus, cervix, and proximal vagina are absent in CAIS because of the action of antimüllerian hormone produced by Sertoli cells of the testis. The vagina varies from a dimple in the perineum to normal length, but is always blind-ending. The treatment options include topical estrogen application, vaginal dilatation, vaginal augmentation techniques, vaginal reconstruction using local tissue flaps, vaginal substitution using colon or artificial tissue engineering materials and preferred timing is prepubertal rather than in early infancy as the patient understanding, participation in decision and co-operation helps achieve optimum results.
Our case presented as familial CAIS and at morbidity meeting, it was discussed in detail. The parents were requested to get the father’s genetic mutation study as the proposed oocyte donor was a known career for the AR gene but this study results were not going to change their plan as they wanted father’s sperm to be used no matter what the AR gene studies indicated. They wanted to do with the budget they have and did not want any additional financial help due to their belief. Our multidisciplinary meeting has noted and respected their decision although was more on the emotional side. Now the rational approach is clearly understood by the parents as they have solution to the problem and have broad joint family so they are not at all affected with the inheritance of CAIS in their goal and taken nothing new approach. Our case is the usual reminder of the fact that underlines the importance of an accurate genetic analysis that has to include karyotype and AR gene variant analysis. This is useful to confirm a clinical diagnosis and establish the proper management of patients with CAIS. With the widespread uptake of non-invasive prenatal testing (NIPT), a larger cohort of women has access to fetal chromosomal sex, which increases the potential to identify prenatal sex discordance. The prenatal diagnosis of androgen insensitivity syndrome (AIS) is an incidental and rare finding [6].
Undescended testes have a small chance of becoming cancerous in the form of gonadoblastoma later in life if they are not surgically removed. Gonadectomy was performed traditionally in childhood as it was thought at increased risk for germ cell cancer called gonadoblastoma (risk <5% in CAIS) and thought that such gonads were without usual hormone function and fertility potential. Accordingly, this risk stratification has for cautious serial observation and/or timing of gonadectomy recommendations to be more individualized and recommend post pubertal gonadectomy or observation for patients with CAIS and strong consideration of prepubertal gonadectomy for those with partial androgen insensitivity syndrome (risk =50%in PAIS) [7]. GCC may take years to become invasive malignancy, which is generally localized and highly curable; thus, observation protocols may be reasonable for select individuals [8]. The estrogen substitutive therapy, if at all indicated as explained above, should be given in the pre-pubertal period at the age of 11 years [9]. Whenever feasible and possible, minimal invasive approach using laparoscopy should be used for bilateral inguinal herniotomy and gonadal biopsies initially at diagnosis in infancy and childhood and during bilateral gonadectomy during adolescence [10,11].
In conclusion, we believe that familial CAIS is very unusual and rare DSDs, and that it is almost impossible to have fertility in the first place and recurrence in the offspring almost unheard of. It is now possible to prevent recurrence in the offspring by taking adequate prenatal investigations and testing and implementing evidence-based approach but in very sensitive case like our initial emotional decision making by parents was respected but they realised the importance of the expert opinion and would now follow the rational approach based on evidence.
Compliance with ethical standards
Conflict of interest
The authors have no conflict of interest to declare. No funding source was involved in this study.
Ethical approval
All procedures performed on human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from the parents and all the relatives involved prior to all the procedures. Parents and all involved parties were informed about the procedure.