Haemangioma is a benign, circumscribed proliferation of miniature, predominantly capillary– sized vascular articulations which are commonly replenished by enlarged blood vessels. Haemangioma is subcategorized into distinct variants. Haemangioma is frequently discerned in infants and young children. An equivalent gender predisposition is observed, especially with variants as synovial haemangioma. Haemangioma can configure as a component of Klippel–Trenaunay syndrome, PHACE syndrome or LUMBAR syndrome [1,2].
Majority of haemangiomas are superficial and situated within head and neck region. Hepatic haemangioma is commonly discerned in infants. Deep seated visceral or skeletal haemangiomas are exceptional, although documented [1,2]. Of obscure aetiology and pathogenesis, haemangioma is contemplated to be a multifactorial lesion associated with endothelial cell proliferation, uncurbed angiogenesis and anomalous functioning of downstream pathways as HIF1α, VEGF and PI3K / Akt [1,2].
Generally, cytogenetic evaluation of haemangioma remains unnecessary. Lesions with RASA1 genetic mutation and association with arteriovenous malformations, Klippel–Trenaunay syndrome, Sturge–Weber syndrome or Parker–Weber syndrome may be challenging to discern. Sturge–Weber syndrome exemplifies GNAQ mutation. Infantile haemangioma is associated with PDGFRβ and downstream signalling pathways as PI3K / Akt / mTOR. Genetic metamorphosis is discerned within intermediate and malignant vascular lesions [1,2].
Classically, superficial haemangioma manifests red to purple papules, nodules or poorly defined, elevated tumefaction [1,2]. Cavernous haemangioma represents as a port wine nevus and may be associated with von Hippel–Lindau disease [1,2]. Visceral haemangioma delineates haemoptysis, visual field defects, localized pressure effect or mass effect. Superficial, cutaneous haemangiomas>5 appear associated with enhanced possible occurrence of visceral haemangioma [1,2]. Hepatic haemangioma is predominantly asymptomatic. Enlarged hepatic haemangioma > 5 centimetres induces abdominal distension and discomfort [1,2]. Preponderantly asymptomatic, skeletal haemangioma may exceptionally depict localized symptoms such as bone pain or pathological fracture. Infantile haemangioma is a miniscule lesion at birth, progresses rapidly up to 6 months and undergoes spontaneous involution following infancy. Congenital haemangioma is maximally enlarged at birth and is devoid of a growth phase. Lesions may expound as partially involuting congenital haemangioma (PICH), completely or rapidly involuting congenital haemangioma (RICH) or may require therapeutic intervention when manifesting as non–involuting congenital haemangioma (NICH) [1,2].
Upon gross examination, deep seated lesions appear well circumscribed, solid to cystic and enunciate distended vascular articulations imbued with or devoid of vascular thrombi. Polypoid tumefaction may depict a definitive stalk [1,2]. Upon microscopy, lobules of capillary–sized vascular channels appear coated with singular layer of flattened, endothelial cells. An enlarged, deep seated, feeding vessel may be discerned. Vascular lobules appear associated with an intervening, stromal lymphocytic infiltrate [1,2]. Haemangioma exhibits distinctive variants denominated as ~anastomosing haemangioma is a deep seated lesion composed of anastomosing vascular channels layered with flattened endothelium.~angiomatosis incriminates multiple tissue planes and enunciates vascular articulations with an irregular, inadequately circumscribed perimeter.~cavernous haemangioma is predominantly configured of ectatic vascular channels [1,2]. ~epithelioid haemangioma is constituted of well formed, miniature vessels coated with plump endothelial cells delineating abundant, eosinophilic cytoplasm and enlarged, spherical nuclei. An extensive, eosinophilic infiltrate appears admixed with vascular articulations. The lobulated, well demarcated neoplasm expounds matured vascular articulations upon tumour periphery [1,2].~glomeruloid haemangioma simulates glomerular capillaries.~hobnail haemangioma is a well circumscribed neoplasm enunciating hobnail nuclei protruding into vascular lumens [1,2].~congenital haemangioma is a solid, rapidly progressive tumefaction with inadequately canalized vascular articulations layered with endothelium demonstrating significant mitotic activity and a circumscribing layer of pericytes. Matured vascular configurations depict prominent lumens with commencement of vascular outflow. Amalgamation of solid and vascular zones is variable(1,2).•non–involuting congenital haemangioma (NICH) exemplifies well configured capillaries and vascular channels(1,2).•involuting congenital haemangioma exhibits vascular articulations with thickened basement membrane and intervening, stromal fibrosis. ~infantile haemangioma delineates proliferating capillary lobules with emergence of distinctive stages as proliferation, partial regression and complete regression [3,4].•early proliferative stage exhibits lobules of immature, dendrite–like cells surrounded by an intervening stroma and an enlarged feeding vessel with occasional perineural involvement [3,4].•early regressive stage demonstrates distension of capillaries with eventual eradication. Basement membrane exhibits accumulated apoptotic debris with elevated peri–capillary mast cells [3,4].•delayed or regression or end stage depicts ghosts of capillaries with circumferential basement membrane and exceptional endothelial cells.Vascular arrangements delineate a phenotype of placental capillaries with immunoreactivity to GLUT1, LeY, CD15, CCR6, IDO or IGF2. ~intramuscular angioma emerges within skeletal muscle admixed with varying quantities of mature adipose tissue, phleboliths and foci of metaplastic bone. Vascular component is constituted of admixed lymphatics, veins of varying magnitude and arteriovenous component. ~lobular capillary haemangioma or pyogenic granuloma exemplifies focal ulceration intermingled with dense, acute and chronic inflammatory exudate [3,4]. ~microvenular haemangioma is inadequately defined and appears confined to superficial and deep seated dermis. Miniature, venule–like channels are layered with singular layer of mitotically inactive endothelial cells surrounded by singular layer of pericytes. The venules lack multi–layering of endothelial cells, traverse through hyalinised dermal collagen bundles and immune non reactive to HHV8 [3,4]. ~sinusoidal haemangioma is a well demarcated proliferation of distended, congested and attenuated vascular channels with miniscule quantities of smooth muscle configuring an anastomosed, sinusoidal tumour pattern. Intervening stroma is scanty. Mitotic activity is absent. ~spindle cell haemangioma is configured of proliferating spindle–shaped cells imbued with intraluminal phleboliths [3,4]. ~verrucous haemangioma exhibits hyperkeratosis and incriminates several tissue planes [3,4]. Although not recommended, ultrastructural examination demonstrates endothelial cells imbued with cytoplasmic folds situated upon luminal surface along with junctional complexes and cytoplasmic pinocytic vesicles (Table 1 & 2) [3,4].
Table 1: Modified International society for Study of Vascular Anomalies [2,3].
Vascular Tumour |
Vascular Malformation |
Infantile haemangioma–focal, segmental, indeterminate |
Slow (low flow) capillary malformations–port wine stain, telangiectasia, angiokeratoma |
Congenital haemangiomas–rapidly involuting congenital haemangioma(RICH), non involuting congenital haemangioma(NICH) |
Venous malformations(VM)– common sporadic VM, Bean syndrome, familial cutaneous and mucosal VM(VMCM) |
Tufted haemangioma |
Glomuvenous malformation (GVM) or glomangioma |
Pyogenic granuloma |
Maffucci syndrome |
Dermatologic acquired vascular tumours |
Lymphatic malformation(LM) –lymphedema, lymphangioma circumscriptum, lymphangioma cavernosum, lymphangioma cysticum |
Kaposiform haemangioendothelioma |
Fast(high flow) malformations–arterial malformation, arteriovenous fistula |
Spindle cell haemangioendothelioma |
Arteriovenous malformation |
Haemangioendothelioma–not otherwise specified (NOS) |
Complex combined vascular malformations |
Table 2: Distinction between congenital and infantile haemangioma (2,3).
Congenital haemangioma |
Infantile haemangioma |
Infrequent (~30%) |
Frequent (~70%) |
Present at birth |
Occurs between 2 weeks to 8 weeks of neonatal period |
Equivalent gender predilection |
Female preponderance (5:1) |
Completely evolved at birth or progresses proportionate to age |
Rapid progression for 6 months to 12 months |
Rapid involution within 12 to 18 months or absence of involution |
Gradual involution |
Immune non–reactive to GLUT1 |
Immune reactive to GLUT1 |
GLUT: glucose transporter protein |
|
Haemangioma is immune reactive to CD31(PECAM1), CD34, ERG, GLUT1, FLI1, von Willebrand factor (vWF), thrombomodulin (CD141), Ulex europaeus agglutinin I(UEAI), claudin5, podoplanin(D2–40), VEGFR3, PROX1, CIZ1 or WTF1(3,4). Haemangioma is immune non reactive to HHV8, TFE3, SMA, desmin or cytokeratin [3,4]. Haemangioma requires segregation from lesions such as granulation tissue, haemangioendothelioma, Kaposi’s sarcoma, angiosarcoma, congenital haemangioma, pyogenic granuloma, Kaposiform hemangioendothelioma, tufted haemangioma, venous malformation, capillary malformation as port wine stain, macrocystic lymphatic malformation and malignant neoplasms as sarcoma or cutaneous sites of neuroblastoma or lymphoma (Figure 1 & 2) [5,6].
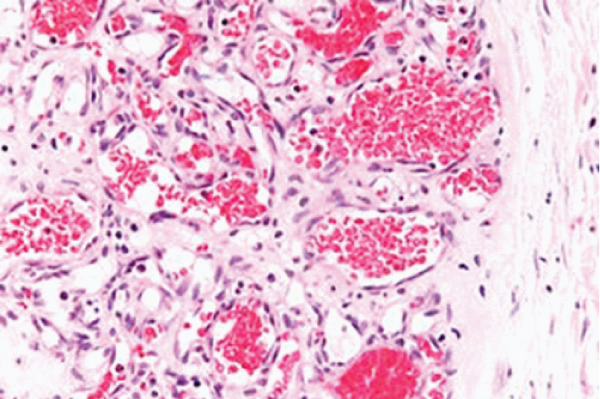
Figure 1: Haemangioma depicting vascular articulations layered with endothelium and imbued with red blood cells surrounded by delicate, fibro–vascular stroma [7].

Figure 2: Haemangioma–cavernous variant depicting large, blood filled spaces layered with endothelium surrounded by delicate, loose, fibrotic stroma [8].
Appropriate discernment of haemangioma requires concurrence of extensive history, physical examination and microscopic features, as morphological countenance is overlapping within diverse clinical representations [5,6]. Rapidly progressive stage of haemangioma manifests transient, mild to moderate thrombocytopenia due to platelet consumption [5,6]. Imaging is predominantly required for assessing extra–cutaneous lesions. Skeletal or bone haemangioma depicts non specific imaging features. Ultrasonography is efficacious and delineates a well defined, extensively vascular tumefaction [5,6]. Contrast enhanced ultrasonography (CEUS) is pathognomonic investigative modality for detecting hepatic haemangioma. Proliferative phase of infantile haemangioma exhibits high vascular flow whereas involuted phase depicts a slow flow. A centric high flow is delineated for NICH haemangioma and slow flow is configured with RICH haemangioma. Grayscale ultrasound adopted for detecting hepatic haemangioma denominates a ‘fluttering sign’[5,6]. Computerized tomography (CT) demonstrates a hypodense lesion with post–contrast enhancement confined to tumour perimeter. Magnetic resonance imaging (MRI) is beneficially employed for discerning deep seated lesions and estimating extent of tissue incrimination. Infantile haemangioma appears hyper–intense upon T2 weighted imaging and isointense upon T1 weighted imaging with post–contrast enhancement [5,6]. Congenital haemangioma exhibits a heterogeneous appearance upon MRI [5,6].
Therapy is directed towards cessation of lesion progression. Contingent to age, site and magnitude, haemangioma is predominantly treated with singular, topical or systemic beta blockers. Besides, corticosteroids, laser therapy or surgical excision may be adopted. Laser techniques, embolization and sclerotherapy or an amalgamation of aforesaid manoeuvers may be employed [5,6]. Contemporary agents as proanthocyanidins appear beneficial as potential modalities for treating infantile haemangioma.
Superficial haemangiomas may undergo ulceration and permanent cutaneous modifications as scarring [5,6]. Localized tumour reoccurrence is uncommon. Reoccurrence may ensue with enlarged, deep seated lesions which are unsatisfactorily treated or inadequately eradicated. Enlarged lesions may contribute to occurrence of high output cardiac failure. Malignant transformation is exceptional [5,6].
None.
The author declares that there is no conflict of interest.